Getting Started with the GPE#
We start with an example of a 1D quantum gas in a periodic potential. In the next document Getting Started in Higher Dimensions, we explore higher dimensions
For our purposes, we consider the following model for a dilute Bose-Einstein condensate (BEC) of neutral \({}^{78}\)Rb atoms in a 1D periodic potential with the Gross-Pitaevski equation (GPE):
Dimensions/Units#
We start by a simple dimensional analysis
To derive others, we note that \(\hbar/m\) and \(g/m\) are independent of mass. These can then be combined to find length and time scales:
Note that this fails in \(d=2\) dimensions and that the 2D GPE famously has a scaling symmetries.
Details
We start with
For distance, we want
For time, we want
Scaling in 2D
In 2D, we have
Thus, we cannot use these to define a distance and length scale. This manifests as an explicit scaling symmetry for solution. To see this, note that if we have a solution \(\psi(\vect{x}, t)\), then the following is also a solution for all \(s\):
The key point is that, under this scaling of dimensions, all terms in the GPE scale the same way by a factor of \(1/s^2\). The overall scaling of \(\psi_s\) follows from its dimensions, which must be \([\psi] = 1/D\) so that the particle number remains fixed:
Before implementing something numerically, we should determine the scales in our problem. We start with a theory perspective: the relevant length scales for this system are:
Density \(n\): The length-scale here is the interparticle spacing, the relation of which to density depends on dimension. In 1D, we have \([n] = 1/L\). This is often expressed in terms of the interparticle spacing \(n^{-1/d}\) where \(d\) is the dimension.
Healing length \(h\): This is the length scale over which the gas will go from the background density \(n_0 = \mu/g\) to zero and represents a balance between the kinetic energy and the non-linear interactions:
\[\begin{gather*} \frac{\hbar^2}{2mh^2} \approx \mu = gn_0, \qquad h = \sqrt{\frac{\hbar^2}{2m\mu}}= \frac{\hbar}{\sqrt{2m g n_0}}. \end{gather*}\]Speed of Sound \(c\): Related to these is the speed of sound. This can be calculated by looking at the Phonon Dispersion from the Linear Response of a homogeneous system to small perturbations:
\[\begin{gather*} c = \sqrt{\frac{gn_0}{m}}. \end{gather*}\]
Equation of State \(\mathcal{E}(n)\).
we have presented everything here for the GPE which models a system with energy density
More generally, we might consider a system that has a different equation of state, and related thermodynamic properties like the chemical potential \(\mu\), pressure \(p\), and the (isoentropic) compressibility \(\kappa\). These are related as follows from standard thermodynamics:
In terms of the equation of state, we thus have
In this sense, the healing length and speed of sound are fundamentally different quantities, and the relationship between the two depends on the nature of the system.
The density \(n\) and healing length \(h\) are intrinsic properties of the system. They represent physics and we must choose relevant scales here. To simulate systems numerically, we must consider some additional length scales:
Box size \(L_x\): This represents the extent of our system and is sometimes called the infrared- or IR-scale* since we cannot simulate features with wavelengths longer than this. Note: this might be physical if we have a trapped gas, where the box size must be larger than the size of the trapped cloud. However, it might also be a numerical artifact when studying homogeneous or periodic systems where we would really like to know what happens in the IR limit (a.k.a. thermodynamics limit) \(L_x \rightarrow \infty\).
Lattice spacing \(\d{x}\): This represents the smallest feature we can resolve in our system, sometimes called the ultraviolet- or UV-scale since we cannot simulate features with wavelengths smaller than this. This is almost never physical, and instead represents a numerical limitation. We almost always want to know what the results are in the UV-limit \(\d{x} \rightarrow 0\).
To reliably simulate physics with our numerical implementation of the GPE we need:
Spatial resolution (UV convergence): The lattice spacing must be smaller than the healing length. When using spectral methods, it often suffices to have \(\d{x}\) a few times smaller. For finite-difference methods, UV convergence is much more difficult to obtain.
\[\begin{gather*} \d{x} < h. \end{gather*}\]IR convergence: The box must be big enough to contain the relevant physics. Together, these set the cost of our simulation – we need \(N_x \geq L_x/\d{x}\) lattice points.
Diluteness: This is not explicitly built into the previous parameters, but the GPE is not valid if the interactions are too strong or if the range of the interaction \(r_0\) is too large. Formally, we must have both
\[\begin{gather*} r_0 \ll n^{-1/d} \ll h. \end{gather*}\]If the healing length is comparable to the interparticle spacing, then there will be an appreciable non-condensed fraction that must be considered, while if the effective range is too large, then the interaction terms \(gn\) must be modified to include three-body interactions etc.
Do It! Canonical Form of the GPE
The GPE can be scaled into a canonical form often seen in the mathematics literature
This is equivalent to choosing a system of units such that \(\hbar = m = g = 1\) so that
but be careful: this does not work in \(d=2\) which has an additional scale invariance. Alternatively, one can change variables:
(Find appropriate constants \(a\), \(b\), \(c\), and \(f\).) One cannot scale away the sign of the interactions. Attractive interactions \(\sigma = -1\) are called focusing while repulsive interactions \(\sigma = 1\) (as we deal with here) are called defocusing. In some of the mathematical literature, you will see this referred to as the (de)focusing non-linear Schrödinger equation (NLSEQ). Various authors might also choose a scaling that absorbs the factor of 2, or includes it with \(\sigma\), and use the acronym (NLS) or (NLSE), so be careful to check the definitions. (Rarely, authors might also change the sign of \(\I\)…)
Question to ponder: What happens to dimensionless parameters such as the diluteness parameter \(an^{1/d}\)?
Note: if the potential is flat \(V(x) = \mu = \) const., then the 1D GPE is integrable, admitting and infinite set of conserved quantities, such as the particle number \(N\), energy \(E\), momentum \(P\), etc. In the dimensionless form, these are related to the constants \(C_i\) defined in Eq.(37) of [Zakharov and Shabat, 1972]:
The first three constants follow from symmetries and Noether’s theorem: \(U(1)\) phase invariance, translation invariance, and time-translation invariance respectively. The rest are a consequence of the integrability.
To enable us to play, we allow the user to set the physical parameters:
healing_length_micron: The healing length \(h\) in microns (micrometers μm = m\(^{-6}\)).dx_healing_length: The lattice spacing in units of the healing length.Lx_microns: The box size in microns.n0_micron: The background/central density in units of inverse microns.
From these, and physical constants such as the particles mass \(m\) and \(\hbar\), we can fix the other physical parameters, such as the chemical potential, and coupling constant.
Defining a Problem#
To set up a simulation, we must create a State class that implements the
gpe.interfaces.IStateGPE interface. Here we define all the parameters in a
subclass. We will also include an external potential, which we will take to be a (periodic)
gaussian
which introduces two new parameters
V0_mu: \(V_0\) expressed in units of the chemical potential \(\mu\).sigma_healing_length: \(\sigma\) expressed in units of the healing length.
Coding Conventions
After some years of experience working with Python code, we have established some coding conventions.
Unless you have an explicit reason not to, follow PEP-8.
Use Black to format your code. This minimizes redundant changes in spaceing, etc. so that version control has less noise and remains useful for exploring significant changes. It also frees developers from agonizing about formatting, improving coding speed.
Most of our code is aimed at high performance in longer simulations. This often requires some 2initialization, precomputing quantities that will be reused etc. before running long simulations. Traditionally, such initialization is done in the contructor (
__init__()in Python), but we would also like to enable users to be able to change attributes or parameters of their simulations after construction. The standard approach is to use getters and setters (@propertyin Python) but dealing with dependencies becomes a chore (i.e. if you change one parameter, you might need update another). After exploring many options, I have landed on a solution that works well for our needs:The constructor sets all attributes with minor checks (types, conversion to arrays etc.), then calls
init()which performs any initializations needed to get the class ready for simulation. Theinit()method will also be called bypre_evolve_hook()andpre_minimize_hook()– part of the intefaces – before simulations/minimization start, allowing changed parameters to be properly considered.We currently provide
*_GPUmethods that are used in the core simulation loops. These return GPU arrays (or other forms of acceleration) and have matching CPU methodsget_Vext()which wrap these and transfer the GPU arrays to the CPU for plotting. (Such transfers are expensive, and should be avoided in the simulations loops.) GPU methods should be carefully designed to allow both CPU and GPU usage for performance. This means thatself.xpshould be used in place ofnpfor example and that all arrays should be assumed to be GPU arrays.Our current recommendation is to use the
Stateto describe the type of system – i.e. Bosons or Fermions with particular interactions or dispersions – and then use anExperimentclass which implementsgpe.interfaces.IExperimentto define the experimental parameters – how is the state prepared, measured, etc.For simplicity, we first define a stand-along state class, then we generalize.
import numpy as np
import matplotlib.pyplot as plt
from gpe.bec import StateGPEBase, u
from gpe.utils import get_good_N
class State(StateGPEBase):
healing_length_micron = 1.0 # Healing length in microns
dx_healing_length = 0.5 # Maximum lattice spacing in units of the healing length
Lx_micron = 10.0 # Box length in units of microns
V0_mu = -1.0 # Depth of the potential in units of the chemical potential.
sigma_healing_length = 1.0 # Width of the potential in healing lengths
n0_micron = 1.0 # Background density
def __init__(self, **kw):
for key in list(kw):
if hasattr(self, key):
setattr(self, key, kw.pop(key))
m = u.m_Rb87
hbar = u.hbar
self.healing_length = self.healing_length_micron * u.micron
Lx = self.Lx_micron * u.micron
dx = self.dx_healing_length * self.healing_length
n0 = self.n0_micron / u.micron
# Select a good Nx that will perform well - small prime factors.
Nx = get_good_N(Lx / dx)
# Calculate the chemical potential and g
mu = hbar**2 / 2 / m / self.healing_length
g = mu / n0
kw.update(hbar=hbar, m=m, Lxyz=(Lx,), Nxyz=(Nx,), mu=mu, g=g)
super().__init__(**kw)
def init(self):
"""Perform any initializations before the simulation begins."""
# Units
self.V0 = self.V0_mu * self.mu
self.sigma = self.sigma_healing_length * self.healing_length
super().init()
def get_Vext(self):
x, = self.get_xyz()
V_ext = self.V0 * np.exp(-(x/self.sigma)**2/2)
return V_ext
s = State()
s.plot();
/home/docs/checkouts/readthedocs.org/user_builds/gpe/conda/latest/lib/python3.14/site-packages/gpe/utils.py:2764: PerformanceWarning: Default State.get_Vext_GPU() delegates to non-GPU version.
This is a potential performance issue, even if not using the GPU. To ensure optimal
performance, you should make sure that you overload get_Vext_GPU() instead.
warnings.warn(
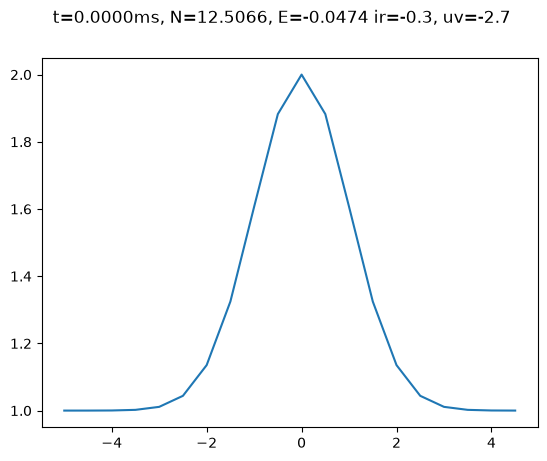
The state is populated using the Thomas-Fermi approximation
We can evolve this and see how close we are to the ground state of the system. Since we want to explore the dynamics, we code this in a function:
from pytimeode.evolvers import EvolverABM
from mmfutils.contexts import FPS
def plot_states(states):
# Always plot dimensionless quantities...
t_unit = u.ms
x_unit = u.micron
n_unit = 1 / x_unit
plot_Nlr = hasattr(states[0], 'get_Nlr')
s = states[-1]
ns = np.array([s.get_density() for s in states])
Es = np.array([s.get_energy() for s in states])
Ns = np.array([s.get_N() for s in states])
if plot_Nlr:
Nlr = np.transpose([s.get_Nlr() for s in states])
ts = np.array([s.t for s in states])
xs = np.ravel(s.xyz[0])
kw = dict(sharex=True, gridspec_kw=dict(hspace=0))
if plot_Nlr:
fig, axs = plt.subplots(3, 1, height_ratios=(0.2, 0.5, 1), **kw)
else:
fig, axs = plt.subplots(2, 1, height_ratios=(0.2, 1), **kw)
ax = axs[0]
E, N = Es[0], Ns[0]
ax.plot(ts / t_unit, Es / E - 1, label="$E$")
ax.plot(ts / t_unit, Ns / N - 1, label="$N$")
ax.set(ylabel="Rel. change", title=f"{E=:.4g}, {N=:.4g}")
ax.legend()
if plot_Nlr:
ax = axs[1]
Nl, Nr = Nlr
ax.plot(ts / t_unit, Nl/Nl[0] - 1, label="$N_l$")
ax.plot(ts / t_unit, Nr/Nl[0] - 1, label="$N_r$")
ax.set(ylabel="Rel. change in $N_{lr}$")
ax.legend()
ax = axs[-1]
mesh = ax.pcolormesh(ts / t_unit, xs / x_unit, ns.T / n_unit)
# Label your axes...
ax.set(ylabel="$x$ [μm]", xlabel="$t$ [ms]")
# ... and your colorbars.
fig.colorbar(mesh, ax=axs.ravel().tolist(), label="$n$ [1/μm]")
def evolve(s, T_ms=20, Nt=200, dt_t_scale=0.4, timeout_s=60):
"""Evolve the state s to time T and plot.
Arguments
---------
s : State
Initial state.
T_ms : float
Final time in ms.
Nt : int
Number of frames to plot.
dt_t_scale : float
Step size for evolve in units of t_scale = hbar/maximum resolvable energy.
timeout_s : float
Timeout (in seconds).
"""
T = T_ms * u.ms
dT = T / Nt
# Note: From experience, if you have chosen dx correctly, you should
# get highly converged results if you use a time-step dt between 0.2 and 0.5 times
# s.t_scale. If you can go much larger, you are probably wasting effort. If need
# to go much smaller, you probably have an incorrect lattice spacing.
dt = dt_t_scale*s.t_scale
# Make sure we can take integer steps...
steps = max(int(np.ceil(dT / dt)), 2)
dt = dT / steps
ev = EvolverABM(s, dt=dt)
states = [ev.get_y()]
for frame in FPS(frames=Nt, timeout=timeout_s):
ev.evolve(steps)
states.append(ev.get_y())
plot_states(states)
return states
states = evolve(s)
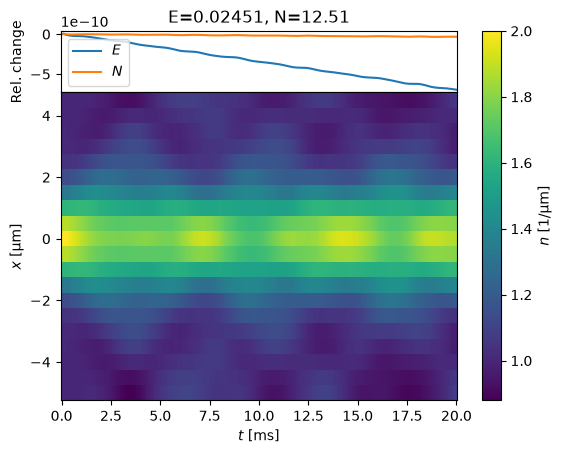
We see some fluctuations, but overall the energy \(E\) and particle number \(N\) are conserved to 9 digits. Here is what might happen if we choose too large of a time-step:
evolve(s, dt_t_scale=1.0);
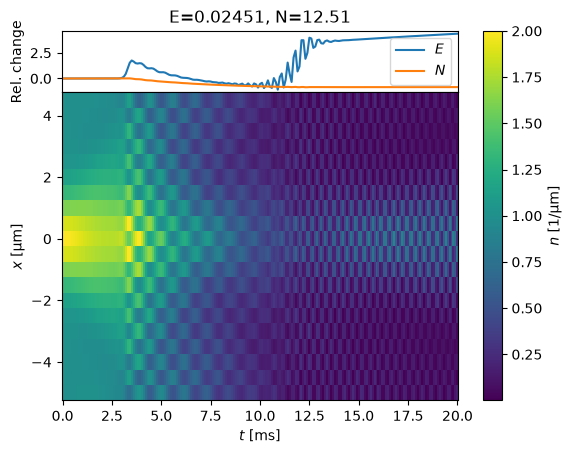
The energy is not conserved here: going larger, the code might crash with ABM.
Minimization#
To get a high-quality initial state, we can run a minimizer:
from gpe.minimize import MinimizeState
s0 = State()
s = MinimizeState(s0, fix_N=False).minimize(use_scipy=True)
evolve(s);
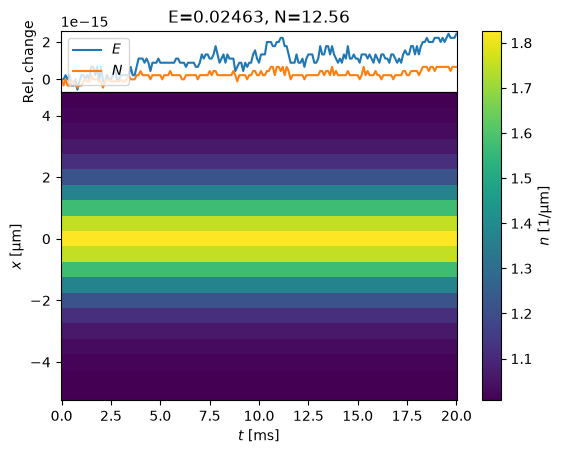
Now the state is static to high precision – the fluctuations that you see are on the order of \(10^{-15}\).
Josephson Oscillations#
Stationary states are pretty boring. Let’s do something more exciting. Here we consider inducing real-space Josephson oscillations as follows:
Place the atoms in a harmonic trap. This needs a new parameter, the trap frequency \(\omega_x\):
\[\begin{gather*} V_{HO}(x) = \frac{m\omega_x^2x^2}{2}. \end{gather*}\]Find the ground state with a repulsive barrier \(V_0 < 0\).
Jump on a linear potential. For a harmonic oscillator, this simply shifts the location of the minimum, so instead, we just use
\[\begin{gather*} V_{HO}(x) = \frac{m\omega_x^2(x-x_0)^2}{2}. \end{gather*}\]
What do you think will happen?
Parameters#
If you are familiar with experiments, you might know what good trap frequencies are, and for a particular species, you can find the mass \(m\) and coupling constant
where \(a_s\) is the S-wave scattering length. Otherwise, it might be better to specify the Thomas-Fermi radius of the trap \(x_{TF}\) where
If we then choose a scale for the central density \(n_0 = g\mu\), this fixes the coupling constant, trap frequencies, etc.
class StateJosephson(State):
x_TF_micron = 50.0
x0_micron = 20.0
Lx_micron = 200.0 # Need bigger box
V0_mu = 2.0 # Repulsive barrier
def init(self):
"""Perform any initializations before the simulation begins."""
# Units
self.x0 = self.x0_micron * u.micron
x_TF = self.x_TF_micron * u.micron
self.wx = np.sqrt(2*self.mu/self.m)/x_TF
super().init()
def get_Nlr(self):
"""Return the number of particles on the left and right respectively."""
x, = self.xyz
n = self.get_density()
N = self.integrate(np.where(x<0, 1, 1j) * n)
Nl, Nr = N.real, N.imag
return Nl, Nr
def get_Vext(self):
x, = self.get_xyz()
V_ext = super().get_Vext()
if (self.initializing or self.t < 0):
x0 = 0.0
else:
x0 = self.x0
V_ext += self.m * (self.wx*(x-x0))**2/2
return V_ext
s0 = StateJosephson()
%time s = MinimizeState(s0, fix_N=False).minimize()
s.plot()
CPU times: user 1.5 s, sys: 5.93 ms, total: 1.5 s
Wall time: 826 ms
/home/docs/checkouts/readthedocs.org/user_builds/gpe/conda/latest/lib/python3.14/site-packages/gpe/utils.py:2764: PerformanceWarning: Default StateJosephson.get_Vext_GPU() delegates to non-GPU version.
This is a potential performance issue, even if not using the GPU. To ensure optimal
performance, you should make sure that you overload get_Vext_GPU() instead.
warnings.warn(
[<Axes: >]
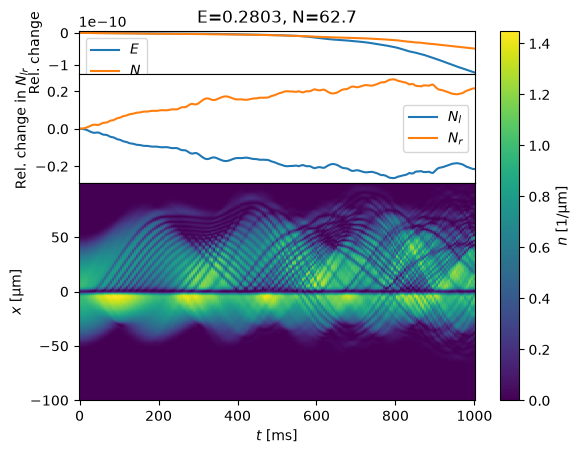
%time evolve(s, T_ms=1000);
CPU times: user 39.8 s, sys: 264 ms, total: 40.1 s
Wall time: 20.2 s
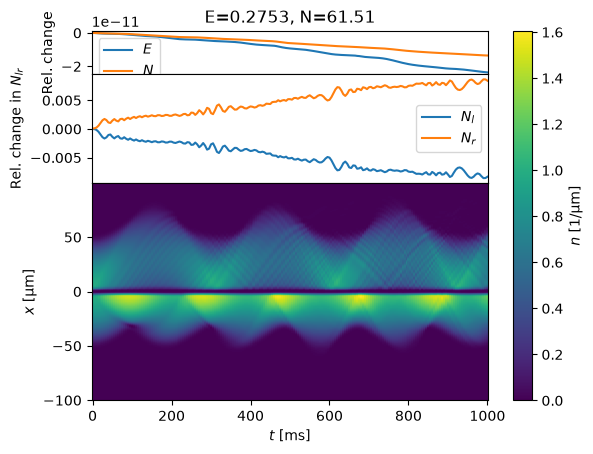
s0 = StateJosephson(V0_mu=4.0)
%time s = MinimizeState(s0, fix_N=False).minimize()
%time evolve(s, T_ms=1000);
CPU times: user 1.07 s, sys: 15 μs, total: 1.07 s
Wall time: 539 ms
CPU times: user 40.4 s, sys: 285 ms, total: 40.7 s
Wall time: 20.5 s
This is fun, but I think we are mixing several different bits of the physics. I suggest you now take the time to review the Josephson effect.
The Josephson Effect
The essence of the Josephson effect is a current
where \(\phi_{-} = \phi_R - \phi_L\) is the phase difference between the BECs on the right and left of the barrier.
DC Josephson Effect#
To generate a DC current, we simply imprint the desired phase difference \(\phi_-\) on the initial state:
class StateJosephsonDC(StateJosephson):
x0_micron = 0.0 # No shift now
dphi = np.pi/2 # Josephson phase difference.
def initialize_state(self):
self.t = 0
s = MinimizeState(self, fix_N=False).minimize(use_scipy=True)
x = s.xyz[0]
phi_x = self.dphi / 2 * np.sign(x)
psi = np.sqrt(s.get_density()) * np.exp(1j*phi_x)
self.set_psi(psi)
s = StateJosephsonDC()
%time s.initialize_state()
%time states = evolve(s, T_ms=200)
CPU times: user 945 ms, sys: 0 ns, total: 945 ms
Wall time: 474 ms
CPU times: user 8.49 s, sys: 59.9 ms, total: 8.55 s
Wall time: 4.33 s
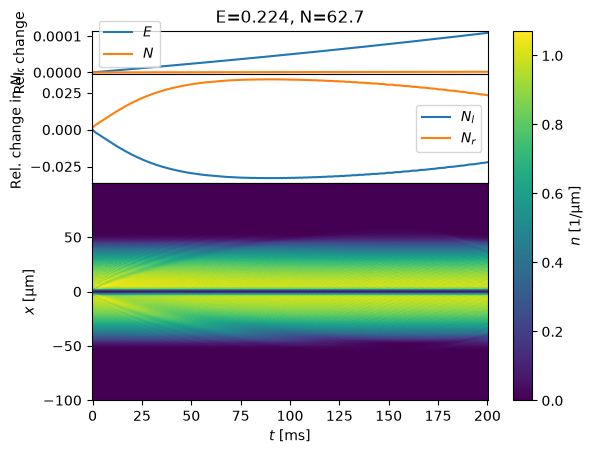
Notice that we start with a constant DC current. After some time, however, this current causes a density difference on either side. The mean-field interaction therefore induces a potential difference \(\delta V \approx g \delta n\) that eventually changes the phase, arresting and reversing the flow.
AC Josephson Effect#
If we instead fix a potential difference difference \(\delta V\), then the phase will wind, generating an alternating current:
class StateJosephsonAC(StateJosephson):
x0_micron = 0.0 # No shift now
dV_mu = 0.2 # Josephson "voltage" difference
def init(self):
self.x0 = self.x0_micron * u.micron
x_TF = self.x_TF_micron * u.micron
self.wx = np.sqrt(2*self.mu/self.m)/x_TF
self.dV = self.dV_mu * self.mu
super().init()
def get_Vext(self):
x, = self.get_xyz()
V_ext = super().get_Vext()
if (self.initializing or self.t < 0):
pass
else:
V_ext += self.dV * np.sign(x)
return V_ext
s0 = StateJosephsonAC()
%time s = MinimizeState(s0, fix_N=False).minimize(use_scipy=True)
%time states=evolve(s, T_ms=1000)
CPU times: user 574 ms, sys: 2 ms, total: 576 ms
Wall time: 308 ms
CPU times: user 41.5 s, sys: 263 ms, total: 41.7 s
Wall time: 21 s
/home/docs/checkouts/readthedocs.org/user_builds/gpe/conda/latest/lib/python3.14/site-packages/gpe/utils.py:2764: PerformanceWarning: Default StateJosephsonAC.get_Vext_GPU() delegates to non-GPU version.
This is a potential performance issue, even if not using the GPU. To ensure optimal
performance, you should make sure that you overload get_Vext_GPU() instead.
warnings.warn(
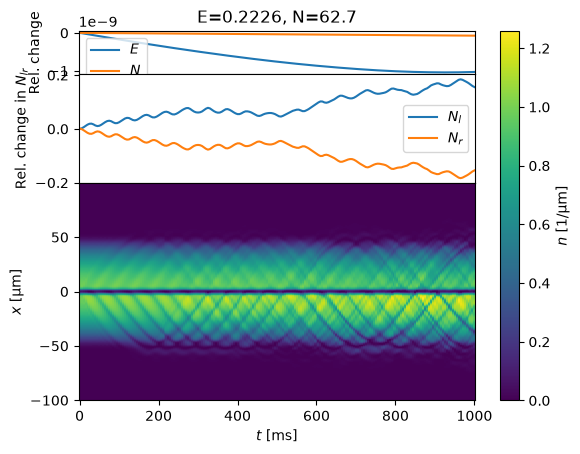
Do it!
Is this the appropriate/expected DC Josephson effect? Check that the corresponding frequencies match. Try playing with the parameters – does the Josephson current respond appropriately?
Note: you might see some deviations from the Josephson formulae. Where might these come from? E.g. what role does the trap play? What about the non-linear interactions? How could you adjust your simulation to test for and minimize these effects?
Finally, how can you adjust the parameters to maximize the signal?